1 min read
Benefits of a Nitrosamine Selective Detector
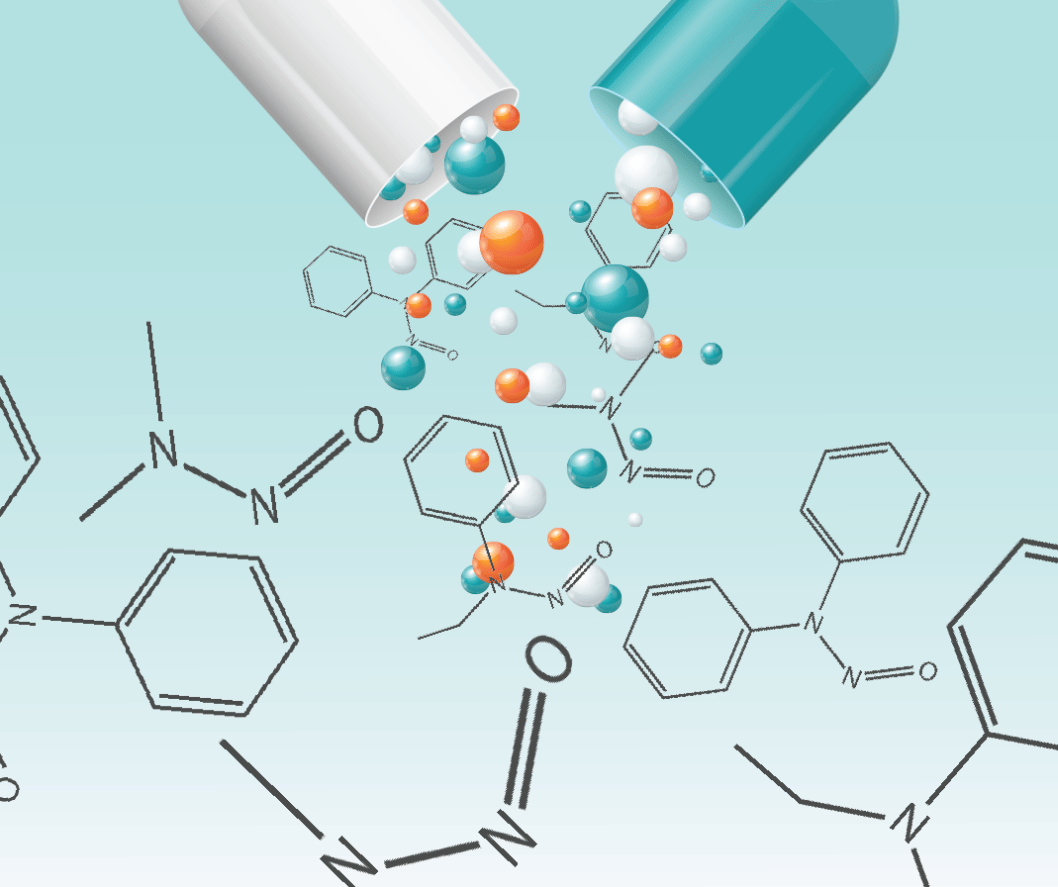
Nitrosamines, such as N-Nitrosodimethylamine (NDMA), are carcinogenic compounds that can form during the drug manufacturing process and storage under...
3 min read
The issue of NDMA contamination in pharmaceuticals like metformin has become an industry-wide focus due to its implications for patient safety and regulatory compliance. NDMA (N-Nitrosodimethylamine) is classified as a probable human carcinogen, and even trace levels can pose significant health risks over time. Regulatory bodies, including the FDA and EMA, have responded with strict guidance for managing and monitoring nitrosamine impurities, urging manufacturers to implement stringent risk assessments, analytical testing, and quality control processes.
This blog provides a detailed overview of NDMA contamination in metformin, discussing why it occurs, how to detect it, and best practices to manage and mitigate risk.
NDMA is a nitrosamine compound that can form as an unintended by-product in pharmaceuticals, particularly during certain synthesis processes. Continuous ingestion of NDMA-contaminated products increases the likelihood of DNA damage, which can result in cancer over time. Recognising this risk, regulatory bodies have set stringent acceptable intake (AI) levels for NDMA to ensure patient health and safety. These limits are determined based on comprehensive toxicological evaluations and lifetime exposure risk models.
NDMA forms when dimethylamine reacts with a nitrating source, such as nitrite, under acidic conditions. Additional risks include nitrite contamination in excipients. Packaging materials, such as nitrocellulose-based components (e.g., blister packs or coatings), can also leach nitrosamines over time, exacerbating contamination risks. Elevated temperatures and high humidity during storage can further accelerate NDMA formation, highlighting the importance of stringent control measures.
To address these risks, manufacturers must evaluate every phase of production for potential contamination points. This includes assessing raw materials, intermediates, excipients, and packaging materials, while monitoring conditions like temperature and pH that may favour NDMA formation. Structured risk evaluation tools, such as the EMA-recommended Failure Mode and Effects Analysis (FMEA), can aid in systematically identifying and mitigating contamination risks.
This is a general formula for the formation of nitrosamines


The FDA, EMA, and other international regulatory agencies have established acceptable intake limits for NDMA, determined by the maximum daily dose and intended treatment duration. These guidelines reflect a unified approach to patient safety but vary slightly in implementation timelines and technical expectations for analytical methods.
Global Challenges: Manufacturers exporting products must align their processes to meet varying international standards, including differing acceptable intake thresholds and validation requirements for analytical methods. For example, while the FDA emphasises product-specific intake limits, the EMA requires a broader lifecycle risk management strategy.

Implement Robust Quality Control Measures: methods, such as Atna for screening, GC-TEA, GC- MS/MS or LC-MS/MS for speciated analysis, should be employed for routine monitoring. Stable isotope-labelled standards can enhance accuracy and mitigate matrix effects during testing.
Conduct Comprehensive Risk Assessments: Manufacturers should use structured tools like FMEA to identify risks in production, storage, and packaging. These assessments must be revisited regularly to reflect updated guidance and evolving risks. By quantitating the nitrite concentration in the excipient facilitates the risk of nitrosamine formation to be properly assessed.
Optimise Production Processes: The synthesise are often well documented and optimised, nitrosamine formation is an issue when amines and nitrosating agents are both present, especially at elevated temperatures or under acidic conditions. Ensuring that solvents used aren’t contaminated with nitrosamines, the excipient being used in the formulation has as low a concentration of nitrites as possible and the inks and packaging materials are free of nitrosamines that can leach into the finished products.
Improving sensitivity and robustness of analytical technique: The use of tools like the ATNA facilitates the high throughput screening of the ingredients, solvent and excipients for the presence of nitrites nitrates and nitrosamines. This allows suspect ingredients to not be used in the formulation, saving time and money. Screening finished product in this way can also rapidly identify samples that require further investigation from products that are immediately safe for release.

In cases where NDMA levels exceed limits, regulatory agencies require immediate action. This includes conducting root-cause analyses, implementing interim measures (such as temporary limits), and revising marketing authorisations. Manufacturers are expected to maintain a robust risk management plan that integrates ongoing monitoring and process optimisation across the product lifecycle.
Lifecycle management strategies should also account for changes in raw material suppliers, new regulatory requirements, and emerging analytical methods.
Controlling NDMA contamination in metformin is vital to safeguarding public health and ensuring regulatory compliance. Utilising cutting-edge detection methods, implementing robust quality control measures, and adhering to FDA and EMA guidelines, pharmaceutical manufacturers can proactively manage NDMA risks. These practices not only ensure safer products but also build trust with patients and regulators alike.

Reference:
FDA Guidance: Control of nitrosamine impurities in Human drugs – Guidance for the industry
https://www.fda.gov/media/141720/download?attachment
EMA guidance: Safeguarding purity under pressure: detecting nitrosamine contamination

1 min read
Nitrosamines, such as N-Nitrosodimethylamine (NDMA), are carcinogenic compounds that can form during the drug manufacturing process and storage under...

1 min read
Nitrosamines are among the most tightly regulated contaminants in pharmaceuticals, food, and tobacco, yet in cannabis, they are barely on the...
1 min read
Nitrosamines have become a pressing concern across industries, particularly pharmaceuticals, food, and environmental safety. These compounds, though...